Desorden neurodegenerativo autosómico dominante, caracterizado clínicamente por desórdenes de movimiento, demencia progresiva, y alteraciones psiquiátricas.
En 1872, George Huntington, MD, presentó la enfermedad como: “corea e impedimento mental de inicio en el adulto y de naturaleza hereditaria, con tendencia al suicidio”.
Esta enfermedad está asociada con una secuencia excesiva de repeticiones CAG en el quinto espacio final del gen HTT (alias IT15) un gen de 350-kD localizado en el brazo corto del cromosoma 4. Los individuos saludables pueden tener entre 9 y 35 repeticiones CAG, mientras que los pacientes diagnosticados con enfermedad de Huntington tienen una anormal expansión de 36 o más repeticiones CAG.
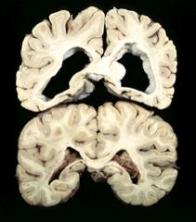
El gen HTT codifica una proteína llamada huntingtin, esta proteína se encuentra neuronas cerebrales, y su función normal se desconoce. En los pacientes afectados, la degeneración neuronal inicia en el cuerpo estriado y progresa hacia la corteza cerebral, ese patrón correlaciona con la progresión demencial de la enfermedad.
Posiblemente la proteína huntingtin anormal es transportada al núcleo, donde sufre agregación, y hace algunas interacciones con otras proteínas explicando la razón de la vulnerabilidad selectiva en estos pacientes.
Neuroquímicamente los niveles de sustancias neurotransmisoras como GABA y su enzima sintética descarboxilada del ácido glutámico están marcadamente disminuidas en los ganglios basales. Los niveles de acetilcolina, sustancia P y encefalinas también se encuentran reducidas. Espectroscopía por resonancia magnética en personas vivas afectadas muestran niveles elevados de lactato en los ganglios basales.
La enfermedad de Huntington se cree puede causar desórdenes psiquiátricos de 2 fromas: Acción directa del gen en las neuronas estriadas, Efecto indirecto del ambiente familiar desordenado en los niños, independientemente de si heredadon o no el gen.
Se creen que entre 5 y 10 personas de cada 100,000 habitantes padecenla enfermedad a nivel mundial. Por su naturaleza progresiva, la enfermedad de Huntington típicamente dura 15 a 20 años desde el inicio de los síntomas hasta la muerte.
Los pacientes afectados más tempranamente (anted de los 20 años), están asociados con ataxia, rigidez, y declinación cognitiva con una progresión mucho más rápida (aprox. 8 años). La edad de aparición usual es entre la cuarta y quinta década de la vida.
Clínica: Los primeros síntomas son movimientos coreiformes típicos (postura distónica y rigidez), o desórdenes psiquiátricos (agresividad, hipersexualidad, impotencia, alcoholismo, psicosis, esquizofrenia, y depresión) mientras que el declive cognitivo general es obvio más adelante.
Tratamiento: Generalmente los inhibidores de la acetilcolinesterasa tienen efectos positivos en el déficit congnitivo, para los demás síntomas las drogas son principalmente para alivio y soporte: Antipsicóticos, antidepresivos, anticonvulsivantes y benzodiacepinas.




No hay comentarios:
Publicar un comentario